Farberin tauti on erittäin harvinainen aineenvaihduntatauti, joka aiheuttaa vakavia fyysisiä heikentymisiä ja johtaa kuolemaan. Vastasyntyneet sairastuvat vain, jos molemmat vanhemmat ovat saman viallisen geenin kantajia. Koska sairaudelle ei ole spesifistä terapiaa, se on tällä hetkellä parantumaton.
Mikä on Farberin tauti?

© phive2015 - stock.adobe.com
Farberin tauti on parantumaton metabolinen sairaus. Tälle sairaudelle on useita nimiä lääketieteessä: Farberin tauti, Farberin oireyhtymä, Ceramodaasin puute tai levitetty lipogranulomatoosi. Farberin oireyhtymä on nimetty amerikkalaisen patologin Sidney Farberin (1903-1973) mukaan, joka löysi taudin ja kuvasi sen.
Tämä sairaus on geneettinen lysosomaalinen sairaus. Lysosomien solunsisäinen häiriö johtuu geneettisestä virheestä, joka on vastuussa haitallisten jätteiden pitkäaikaisesta varastoinnista kehossa. Koska maailmanlaajuisesti on vain muutama Farber-potilas, tautia ei vieläkään tunneta hyvin. Potilaat kuolevat yleensä lapsenkengissä, mutta enimmäkseen ennen kolmen vuoden ikää.
syyt
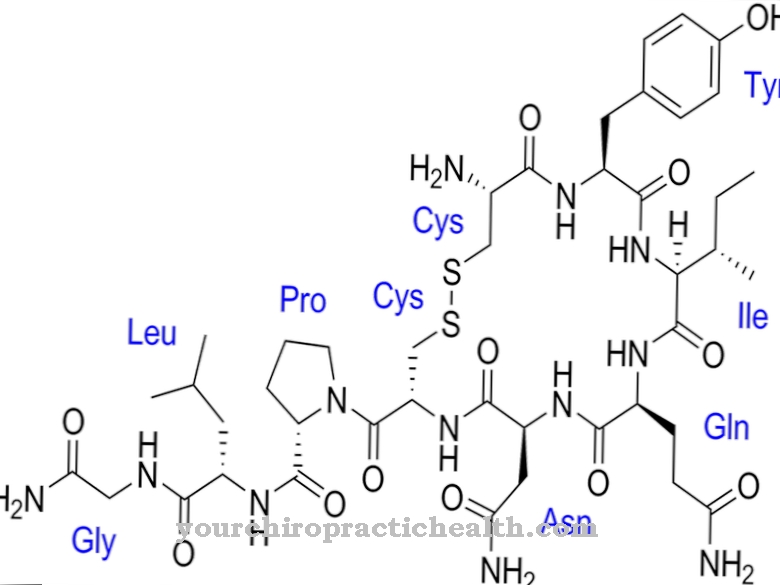
Farberin tauti johtuu ASAH-geenin mutaatiosta. Mutaatio välittyy autosomaalisesti recessiivisesti vanhemmilta sukupolvilta jälkeläisille. ASAH-geeni koodaa entsyymejä keramidaasia ja happokeramidaasia. Kuitenkin, jos geeni mutatoituu, keramidaasin ja happokeramidaasin aktiivisuus puuttuu. Happo keramidaasi on entsyymi, joka vastaa lysosomaalisesta hydrolaasista.
Hydrolaasi on katalyysi, johon osallistuu keramiidaasi-entsyymi, jossa lipidikeramidi jaetaan aminoalkoholisfingosiiniksi ja rasvahapoksi. Farber-potilailla tämä prosessi on häiriintynyt. Tällä tavalla lähtöaine, keramiidi, pysyy jakamattomana soluissa ja sitä varastoidaan siellä pitkään. Alkuperäisessä tilassaan oleva keramidi on keholle jätettä, koska sitä ei voida jalostaa tarvittavaksi lopputuotteeksi geneettisen vian takia.
Siksi Farberin oireyhtymä on aineenvaihduntatauti. Aineenvaihduntahäiriöiden seurauksena voi ilmetä erilaisia valituksia, jotka vaihtelevat potilaittain. Myös Farberin taudissa ajankohta, jolloin taudin merkit ilmenevät, on hyvin erilainen: joillakin potilailla ensimmäiset oireet ilmenevät jo lapsuudessa, toisissa kuitenkin vain vastasyntyneissä. Harvinaisissa tapauksissa tauti ei tunne itseään ennen murrosiän alkamista.
Löydät lääkkeesi täältä
Pain KivulääkkeetOireet, vaivat ja oireet
Taudin kulku on hyvin erilainen. Kun ensimmäiset sairauden merkit ilmestyvät lapsenkengissä, ne ilmenevät yleensä ensin motorisen häiriön muodossa. Esimerkiksi vauvat eivät voi venyttää raajojaan tai jalkojaan ja ovat hyvin rajoitetusti liikkuvissa.
Nivelkontraktiot voivat myös aiheuttaa kipua, joka vaatii kipulääkehoitoa. Lisäksi periartikulaariset kyhmyt voivat muodostua elimissä tai nivelissä hyvin varhain. Solmut, jotka ovat vain ihon alla, näkyvät myös paljain silmin. Imeväisillä kurkunpään muutos on myös tyypillinen Faberin taudille.
Kurkunpään muutoksen yleinen termi on laryngomalacia. Pahimmassa tapauksessa kurkunpään muutos johtaa hengitysteiden kaventumiseen hengitystieinfektioiden kanssa ja riittämättömän hapen saannin kanssa, samoin kuin vaikeaseen ruoanottoon kasvuhäiriöillä. Lyhyt kasvu on kuitenkin tyypillinen merkki Faberin taudista riippumatta laryngomalaciasta.
Sarveiskalvon opasiteetti ja neurologiset poikkeavuudet ovat muita oireita Faberin oireyhtymästä. Ei ole harvinaista, että perna ja maksa suurenevat. Hepatosplenomegalia on, kun molemmat elimet suurenevat samanaikaisesti. Jos sairaus etenee aggressiivisesti tai jos diagnoosi viivästyy, kuolema voi tapahtua jo ensimmäisen elämän vuoden aikana.
diagnoosi
Farberin oireyhtymä voidaan löytää ja diagnosoida erittäin nopeasti. Diagnoosi tehdään joko mittaamalla keramidaasin entsyymiaktiivisuus tai mittaamalla keramidin hajoaminen. Keramidin hajoaminen tapahtuu valkosoluissa tai viljellyissä ihon fibroblasteissa. Epäilyttäessä voidaan myös tehdä synnytyksen diagnostiikka, esimerkiksi jos tauti tunnetaan esi-isässä.
komplikaatiot
Useimmat Farberin taudin tapaukset johtavat kuolemaan. Taudin hoito ei ole mahdollista. Taudin aikana syntyvät komplikaatiot vaihtelevat suuresti. Oireita esiintyy kuitenkin varhaisessa lapsuudessa, joten potilaat eivät voi enää liikuttaa raajojaan kunnolla.
Arkielämässä on vakavia rajoituksia. Potilas on riippuvainen muiden ihmisten avusta. Monissa tapauksissa esiintyy myös kipua, vaikka sitä voidaan hoitaa kivuhoidon avulla. Kurkunpään muutokset lisäävät hengitystieinfektioiden riskiä, joka voi johtaa kuolemaan.
Lyhyt vartalo esiintyy myös voimakkaammin Farberin taudissa ja vaikeuttaa potilaan elämää. Maksa ja perna voivat myös olla laajentuneet. Taudin hoito ei ole mahdollista. Useimmissa tapauksissa kipulääkkeitä käytetään tekemään arjesta sietävä.
Epämuodostumien tai muodonmuutosten tapauksessa voidaan käyttää kirurgisia toimenpiteitä, joilla sairaus lievitetään. Enemmän komplikaatioita ei ole. Luuytimensiirrot voivat myös vähentää oireita, mutta Farberin taudista ei ole täydellistä paranemista.
Milloin sinun pitäisi käydä lääkärillä?
Valitettavasti Farberin tautia ei voida hoitaa tai estää suoraan. Loppujen lopuksi se johtaa lapsen kuolemaan ennen kuin lapsi on edes vuoden ikäinen. Lääkärin kanssa on neuvoteltava, jos lapsi kärsii motorisista ja psyykkisistä vammoista. Myös lapsen liikkuminen on rajoitettu merkittävästi.
Vakavan kivun takia monet lapset huutavat jatkuvasti ja ihon alle muodostuu pieniä kohoumia. Lääketieteellinen hoito on välttämätöntä myös hengitysteiden infektioille. Nämä ovat havaittavissa lapsen hengittäessä voimakkaasti. Myös kehitys ja kasvu ovat lyhyitä ja yleisiä häiriöitä.
Farberin taudin voi diagnosoida yleislääkäri tai lastenlääkäri. Eri asiantuntijat suorittavat kuitenkin lisähoitoa. Lisäksi monet vanhemmat ja sukulaiset ovat riippuvaisia psykologisesta hoidosta. Tätä voidaan pyytää myös suoraan sairaalasta. Jos Farberin taudin oireet vaikuttavat akuuteilta ja vaarantavat lapsen hengen, on joka tapauksessa kutsuttava ensiapu.
Lääkärit ja terapeutit omalla alueellasi
Hoito ja hoito
Farberin taudille ei vielä ole tehokasta terapiaa. Oireet hoidetaan oireellisesti kipulääkkeillä ja glukokortikoideilla. Viimeksi mainitut estävät tulehduksellisia reaktioita ja auttavat hallitsemaan solujen metaboliaa. Plastiikkakirurgian avulla kehon vakavat muodonmuutokset voidaan korjata. Tällä tavalla oireita lievitetään.
Hoidot helpottavat potilaiden elämää jossain määrin. Tauti etenee kuitenkin ajan myötä. Tällä hetkellä luuytimensiirto lupaa lievittää ja parantaa tautia. Maailmanlaajuisesti tunnetaan 50 tapausta, joissa luuytimensiirto suoritettiin onnistuneesti. Vielä on epäselvää, pystyykö lääke parantamaan tautia pitkällä tähtäimellä.
Näkymät ja ennuste
Farberin taudin ennuste on erittäin huono. Tauti esiintyy vain hyvin harvinaisissa tapauksissa, mutta johtaa jokaisella toistaiseksi dokumentoidulla potilaalla varhaiseen kuolemaan. Vaikuttavilla potilailla on vanhempia, joilla molemmilla on sama geneettinen virhe. ASAH-geeni on mutatoitunut sekä biologisessa isässä että biologisessa äidissä.
Tämä tarkoittaa, että viallinen geeni siirtyy kohdussa syntymättömän lapsen kehitysprosessin aikana. Vastasyntynyt lapsi kärsii väistämättä vakavasta aineenvaihdunnasta. Häiriö on parantumaton riittämättömien lääketieteellisten vaihtoehtojen takia.
Tutkijoiden ja tiedemiesten ei tällä hetkellä sallita puuttua ihmisen genetiikkaan laillisista syistä. Tämä pätee myös vakaviin sairauksiin. Siksi tutkitut ja testatut lääkkeet, hoidot ja hoitomenetelmät perustuvat potilaan oireisiin. Kaikista ponnisteluista huolimatta toistaiseksi ei ole olemassa riittävää lääketieteellistä hoitoa tai vaihtoehtoisia parantamismenetelmiä, jotka johtavat oireiden vähentämiseen tai olemassa olevien oireiden pitkäaikaiseen lievittämiseen.
Farberin tautia sairastavat potilaat kuolevat elämänsä ensimmäisten vuosien aikana. Koska useimmissa tapauksissa kuolema tapahtuu kolmannella elämänvuonna, on epätodennäköistä, että kuolemat saavuttavat esiopetuksen.
Löydät lääkkeesi täältä
Pain Kivulääkkeetennaltaehkäisy
Farberin tautia ei voida estää, koska tauti periytyy autosomaalisesti taantuvalla tavalla. Autosomaalinen taantuva perintö tarkoittaa, että molemmilla vanhemmilla on oltava mutatoitunut geeni lapsen sairastumiseksi. Lisäksi viallisen alleelin on oltava molemmissa homologisissa kromosomeissa. Juuri näistä syistä tauti on niin harvinainen.
Ja se voi ohittaa useita sukupolvia, kunnes vastasyntynyt perheenjäsen sairastuu uudestaan. Faber-tautipotilaiden jälkeläisillä taudin todennäköisyys on noin 25 prosenttia. Vaikka vanhemmat voivat siirtää taudin lapsille geneettisen vian vuoksi, he ovat yleensä itse terveitä.
Ja jos vain yhdellä vanhemmilla on viallinen geeni, omat lapsesi pysyvät terveinä, mutta he voivat siirtää viallisen geenin jälkeläisilleen.
Jälkihoito
Munasarjojen syövän seurannassa hoidon päättymisen jälkeen keskitytään kasvaimen uusiutumisen havaitsemiseen, hoidon sivuvaikutusten seurantaan ja hoitoon, psykologisiin ja sosiaalisiin ongelmiin kärsivien potilaiden tukemiseen sekä elämänlaadun parantamiseen ja ylläpitämiseen.
Hoidon jälkeen suositellaan tarkistuksia gynekologin kanssa kolmen kuukauden välein. Se, kuinka kauan tarkastukset ovat välttämättömiä, riippuu hoitavan lääkärin arviosta. Yleensä gynekologi aloittaa tarkistuksen yksityiskohtaisella keskustelulla, jossa fyysisten valitusten lisäksi psykologiset, sosiaaliset ja seksuaaliset ongelmat ovat merkityksellisiä.
Sen jälkeen gynekologi suorittaa yleensä gynekologisen tutkimuksen ja ultraäänitutkimuksen. Potilaat, joilla ei ole erityisiä oireita, eivät tarvitse lisäselvityksiä. Jos oireita ilmenee ajan myötä, kuten vatsan koon lisääntyminen vedenpidätyksen tai hengenahdistuksen vuoksi, lisätutkimukset, kuten CT, MRI tai PET / CT, voivat olla hyödyllisiä.
Hoidon aikana ilmenevät valitukset tulisi ottaa vakavasti potilaiden puoleen, josta niistä on keskusteltava hoitavan gynekologin kanssa. Munasarjasyövän hoitoon sisältyy usein radikaalia leikkausta. Siksi tarkastuksia olisi käytettävä operaation mahdollisten seurausten tunnistamiseen ja hoitamiseen varhaisessa vaiheessa. Tarvittavan kemoterapian mahdollisia sivuvaikutuksia voidaan myös seurata säännöllisillä tarkastuksilla.
Voit tehdä sen itse
Faberin tauti on hyvin harvinainen perinnöllinen aineenvaihduntahäiriö, jota pidetään parantumattomana ja johtaa usein lapsuudessa kuolemaan. Koska tauti on geneettinen, sairastuneet eivät voi ryhtyä omaehtoisiin toimenpiteisiin, joilla on syy-vaikutus.
Pari, jonka perheissä Faber-oireyhtymä on jo tapahtunut, voi kysyä perintöneuvontaa ennen perheen perustamista. Osana tätä kuulemista sinulle ilmoitetaan todennäköisyydestä, että jälkeläiset kärsivät tästä häiriöstä, ja mihin stressiin heidän on varauduttava tässä tapauksessa.
Hyvin usein Faberin tautia sairastavat lapset kuolevat vauvoina. Asianomaisten vanhempien on odotettava, että heidän lapsensa ei saavuta kolmen vuoden ikää. Perheiden ei pidä kantaa tätä valtavaa tunnetaakkaa yksin. Sukulaisia suositellaan ottamaan yhteyttä psykoterapeuttiin diagnoosin jälkeen. Monet vanhemmat hyötyvät myös vaihtoista muiden samanlaisessa tilanteessa olevien perheiden kanssa.
Koska Faberin tauti on hyvin harvinainen, erityisiä tukiryhmiä ei ole. Vakavassa syövässä olevien lasten sukulaiset ovat alttiina samanlaisille stressille. Tällaisiin ryhmiin kuuluminen voi siksi olla hyödyllinen päivittäisten stressien selvittämisessä ja selviytymisessä paremmin muiden kärsivien henkilöiden avulla.
.jpg)
.jpg)





.jpg)

.jpg)





.jpg)