Entsyymikorvaushoito sitä käytetään hoitamaan lysosomaalisia varastointitauteja, joissa entsyymien puute johtaa hajoamistuotteiden patologiseen kertymiseen solujen lysosomeihin.
Geneettisistä virheistä johtuvat puuttuvat entsyymit kompensoidaan säännöllisillä suonensisäisillä infuusioilla. Koska infusoidut synteettiset entsyymit eivät voi molekyylikokoonsa mennessä ylittää veri-aivoestettä, hoito toimii vain sellaisten lysosomaalisten varastointitautien hoidossa, jotka eivät vaikuta keskushermostoon.
Mikä on entsyymikorvaushoito?

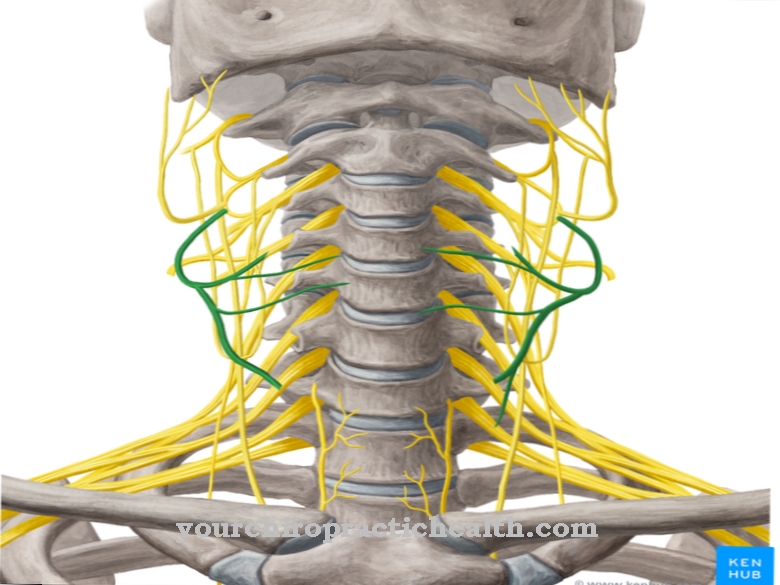
Lysosomit ovat erityisiä soluorganelleja, joissa vieraat ja endogeeniset aineet hajoavat ja kierrätetään osittain. Erityisiä hydrolysoivia entsyymejä tarvitaan aineiden hajoamiseen ja kuljettamiseen. Nämä ovat proteaaseja, nukleaaseja, lipaaseja ja kuljetusaineita.
Lukuisat tunnetut geneettiset viat voivat johtaa tiettyjen entsyymien vajaatoimintaan, joten osa hajoamistuotteista kertyy lysosomeihin patologisina määrinä ja kertyy, kunnes ne saavuttavat solunulkoisen matriisin, ts. Solujen väliset tilat, hallitsemattomasti. Kaikki geneettiset viat, jotka johtavat ainakin yhden välttämättömän hydrolaasin vajaatoimintaan, on yhteenveto termillä lysosomaalinen varastointitauti. Entsyymikorvaushoito (ERT, entsyymikorvaushoito) käytetään korvaamaan puuttuvat endogeeniset entsyymit synteettisesti valmistetuilla entsyymeillä.
Koska hydrolaasit koostuvat suhteellisen suurista molekyyleistä, niitä ei voida imeytyä suolistosta ensin hajottamatta ja inaktivoimatta, joten ne voidaan antaa vain laskimonsisäisenä infuusiona. Entsyymimolekyylien koko estää kuitenkin myös veri-aivoesteen ylittymisen, joten terapia voi olla tehokas vain sellaisten lysosomaalisten varastointitautien varalta, jotka eivät vaikuta keskushermostoon (CNS).
Toiminta, vaikutus ja tavoitteet
Tunnetaan yli 50 erilaista lysosomaalista aineenvaihduntahäiriötä, joista jokainen voidaan jäljittää monogeneettiseen vikaan. Lysosomaaliset varastointitaudit voidaan jakaa seitsemään eri luokkaan riippuen liiallisesti varastoiduista aineista olemassa olevan entsyymivian vuoksi.
Mukopolysakkaroidit ja oligosakkaridoosit soveltuvat ensisijaisesti ERT: hen. ERT: n tavoitteena on aina kompensoida spesifisen entsyymin puutos keinotekoisesti toimitettujen entsyymien avulla taudin pysäyttämiseksi tai ainakin lievemmäksi kulkemiseksi. Yksityiskohtaisesti, korvaavia entsyymejä on saatavana seuraaviin lysosomaalisiin varastointitauteihin:
- Gaucherin tauti
- Pompe-tauti
- Fabry-tauti
- Hurler-Pfaundler-oireyhtymä (mukopolysaccharidosis I)
- Hunterin tauti (mukopolysakkaridioosi II)
• Maroteaux-Lamy-oireyhtymä (mukopolysaccharidosis VI) • Niemann-Pick B
Gaucherin tauti on yleisin lysosomaalinen varastointitauti. Sitä esiintyy kolmessa eri variantissa, joista kaksi vaikuttaa myös hermostoon. Ei-neuropaattisessa muodossa pernaan kohdistuu erityistä huomiota, mikä suurenee suuresti ja johtaa sekundaarisiin vaurioihin, kuten anemiaan ja luuytimen vaurioihin. Tyypillisiä oireita ovat luu- ja nivelkipu ja verenkiertohäiriöt. Taudin akuutti neuropaattinen variantti osoittaa vaikean kulun ja tarjoaa vähän mahdollisuuksia selviytymiseen kahden ensimmäisen elämävuoden jälkeen.
Varastointitauti Pompe-tauti johtuu alfa-1,4-glukosidaasi-entsyymin puutteesta, joka on mukana monissa aineenvaihduntaprosesseissa. Pompe-tauti johtaa sydämen valtavaan laajentumiseen (kardiomegalia) ja sydämen vajaatoimintaan. On varhaisia, vakavia kursseja, jotka ilmestyvät muutaman ensimmäisen elämän kuukauden aikana, sekä lievempiä muotoja, jotka ilmestyvät vasta myöhemmin elämän vuosina.
Fabry-tauti johtuu X-kytketystä geneettisestä vikasta, joten säilytystauti voi vaikuttaa vain poikiin ja miehiin. Tauti johtaa yleensä pitkälle edenneessä lapsuudessa esiintyviin oireisiin, mukaan lukien kipuhyökkäykset, ihon keratoomat, munuaisongelmat ja sydänlihavauriot. Alfa-galaktosidaasi A -entsyymin puute johtaa keramiditriheksosidin kertymiseen, mikä aiheuttaa oireita, jotka voivat myös vaikuttaa autonomiseen hermostoon.
Ei ole harvinaista, että vauriot johtavat sydänkohtaukseen, munuaisten infarktiin tai jopa aivohalvaukseen. Hurler-Pfaundler-oireyhtymä tunnetaan myös tyypin I mukopolysakkharidoosina, ja sen aiheuttaa glykosaminoglykaanimetabolian häiriö. Tauti liittyy moniin erilaisiin oireisiin, mukaan lukien vaikea henkinen vajaatoiminta ja vakavat luuston muutokset. Taudin kulku on vakava, joten keskimääräinen elinajanodote on 11–14 vuotta. Hunterin tauti vastaa tyypin 2 mukopolysakkharidoosia, ja sen aiheuttaa - kuten Hurlerin tauti - X-kytketty vika. Taudille on ominaista vaihtelevan vakavuusaste, varhaislapsuudesta lieviin, vain aikuisilla miehillä esiintyviin kursseihin.
Yleisimmistä sydämen oireista, kuten sydämen venttiilin vikoista ja sydänlihaisongelmista johtuen elinajanodote vaihtelee normaalista lievästi rajoitettuun. Maroteaux-Lamy-oireyhtymä (MPS VI) on yksi mukopolysakkaridista, joka periytyy autosomaalisesti recessiivisella tavalla, koska syy-geenivika ei ole X-kromosomissa. Tauti on hyvin harvinainen, yksi tapaus 455 000 syntymää kohden. On tunnettuja lieviä ja vakavia muotoja.
Oireita ovat laajentunut maksa ja perna, karpaalitunnelin oireyhtymä ja muutokset sydänventtiileissä. Niemann-Pick B on sfingomyeliinin lipidoosi, joka on yksi lysosomaalisista varastointitaudeista ja johtuu kromosomin 11 geenivirheestä. Vaikka taudin tyyppi B vaikuttaa pääasiassa maksaan ja pernaan, tyypillä A on myös huomattavia hermosto-ongelmia.
Löydät lääkkeesi täältä
Pain KivulääkkeetRiskit, sivuvaikutukset ja vaarat
Koska monet lysosomaalisista varastointitauteista, joita voidaan hoitaa entsyymikorvaushoidolla, vievät vakavaan kurssiin vastaavasti korkeamman kuolleisuuden, jos niitä jätetään hoitamatta, ERT: n suurin riski on, että valittu korvaava entsyymi ei toimi tai toimii vain liian heikosti.
Toinen riski on vähemmän itse terapiassa kuin siinä, että perussairaus tunnistetaan liian myöhään, jotta ERT voi pysähtyä hoidon aikana, mutta jo aiheutetut vauriot eivät voi taantua. Noin joka toinen hoidettu väliaikaisesti potilas reagoi infuusioihin oireilla, kuten kuume ja vilunväristykset. Syitä tähän ei ole vielä täysin ymmärretty. Jotkut potilaat reagoivat muodostamalla vasta-aineita, ja on ollut tiedossa tapauksia, joissa potilaat ovat reagoineet ihottumien ja bronkospasmin kanssa.



.jpg)






.jpg)