lysosomeihin ovat organelleja elävien asioiden soluissa, joissa on kehittyneitä solun ytimiä (eukaryootteja). Lysosomit ovat solun vesikkeleitä, joita ympäröi kalvo ja jotka sisältävät ruuansulatusentsyymejä. Hapanta ympäristössä pidettyjen lysosomien tehtävänä on hajottaa endogeenisiä ja eksogeenisiä aineita ja tarvittaessa käynnistää solujen tuhoaminen (apoptoosi).
Mikä on lysosomi?
Lysosomit ovat vesikkeleitä, ts. Eukaryoottisolujen pienten solujen sulkeumia, joita ympäröi kalvo ja jotka sisältävät erilaisia solunsisäisiä, hydrolyyttisiä ruuansulatuksen entsyymejä. Nämä ovat proteaaseja, nukleaaseja ja lipaaseja, ts. Ruoansulatusentsyymejä, jotka voivat hajottaa ja hajottaa proteiineja, nukleiinihappoja ja rasvoja.
Fragmentit joko hajoavat edelleen, ja ne hävitetään osittain tai metaboloidaan uudelleen, niin sanottuna kierrätetään. Lysosomeja kutsutaan siksi myös solun omaksi vatsaksi. Lysosomien sisätilat, joiden halkaisija on 0,1 - 1,1 mikrometriä, pidetään happamassa väliaineessa, jonka pH on 4,5 - 5,0, protonipumppujen aktiivisuuden avulla. Voimakkaasti hapan ympäristö suojaa itse solua, koska entsyymit toimivat vain happamassa ympäristössä.
Jos lysosomi tyhjentää entsyyminsä pH-neutraaliin sytosoliin, ne deaktivoituvat välittömästi ja ovat vaarattomia solulle. Jotta ruuansulatusentsyymit eivät hyökkää itse kalvoon, membraaniproteiinit glykosyloidaan voimakkaasti sisäpuolelta.
Toiminto, vaikutukset ja tehtävät
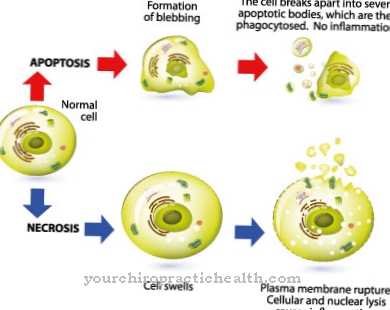
Lysosomien päätehtävänä on tarjota hydrolyyttisiä ruuansulatusentsyymejä proteiinien, nukleiinihappojen ja rasvojen hajottamiseksi tarvittaessa. Ne voivat olla aineita, jotka ovat vieraita solulle tai omille. Soluaineiden hajoaminen sisältää myös apoptoosin, esiohjelmoidun solukuoleman, jossa lysosomit entsyymiensä kanssa suorittavat olennaisen teknisen toiminnan.
Solun ulkopuoliset partikkelit, jotka ovat solunulkoisessa tilassa ja jotka on tarkoitettu hajoamiseen, kuljetetaan ensin soluun endosytoosin avulla. Ulompi solukalvo kääntyy ulospäin, virtaa hajottavan aineen ympäri ja halkeilee sitten solukalvosta itsenäisenä rakkuna. Vesikkelit sulautuvat lysosomeihin siten, että hajoamisprosessi voi alkaa. Endosytoosin prosessi ja fuusio lysosomin kanssa tapahtuu aina ilman suoraa kosketusta sytoplasmaan ja on verrattavissa fagosytoosiin.
Osana itsenäistä solujen uusimisprosessia myös muut sytosolin organelit ja komponentit syötetään lysosomeihin ”hajoamista” varten. Yleensä fragmentteja käytetään uudelleen aminohappojen, proteiinien, nukleiinihappojen ja hiilihydraattien rekonstruointiin, ts. Kierrätetään. Lysosomilla on myös tärkeä rooli apoptoosissa, ohjelmoidussa solukuolemassa. Apoptoosisignaalin vastaanottanut solu kutistuu ja puretaan tietyn ohjelman mukaisesti ilman, että solun osat joutuvat solunulkoiseen tilaan, missä tulehdukselliset reaktiot tapahtuisi välittömästi.
Koulutus, esiintyminen, ominaisuudet ja optimaaliset arvot
Lysosomeja esiintyy käytännössä hyvin harvoin poikkeuksin eukaryoottien jokaisessa solussa. Vain lysosomien lukumäärä solua kohden vaihtelee solutyypistä ja solun tehtävistä kudoksessa. Lysosomikalvon hydrolyyttiset entsyymit ja proteiinit syntetisoidaan ribosomien avulla endoplasmisessa retikulumissa (ER). Sitten ne merkitään trans-Golgi -laitteeseen siten, että niitä ei kuljeta sattumanvaraisesti mihinkään lysosomeihin.
Tärkein merkitys merkinnöissä on fosfotransferaasilla ja toisella entsyymillä, joka täydentää merkintöjä. Lysosomien happaman ympäristön varmistaa V-tyypin ATPaasi. Entsyymi halkaisee 2 H + -ionit ATP: stä hydrolyysiprosessin kautta ja kuljettaa ne lysosomiin. Lysosomit osallistuvat lukuisiin sisäisiin ja ulkoisiin aineenvaihduntaprosesseihin. Heidän lukumäärän suora tai epäsuora mittaus ei ole mahdollista, ja sillä olisi vähän informatiivista arvoa. Siksi ei voida lausua optimaalisesta määrästä lysosomeja. Mahdolliset lysosomien toimintahäiriöt ovat yleensä erittäin havaittavissa.
Sairaudet ja häiriöt
On olemassa useita lysosomien toimintahäiriöitä, jotka johtavat vakavaan sairauteen. Hyvin harvoin esiintyvä - geneettinen - toiminnallinen häiriö laukaistaan fosfotransferaasin puutteella. Toimimaton entsyymi johtaa lysosomaalisten entsyymien hallitsemattomaan vapautumiseen solunulkoiseen matriisiin.
Samaan aikaan lysosomeihin kertyy lipidejä, mukopolysakkarideja ja glykoproteiineja, jotka on tosiasiallisesti tarkoitettu hajoamiseen ja hajoamiseen. Mutta koska ruoansulatusentsyymejä ei ole niiden väärän suunnan takia, aineet kerääntyvät yhä enemmän lysosomeihin. Tämä autosomaalinen, recessiivinen peritty lysosomaalinen varastointitauti, joka tunnetaan nimellä I-solutauti, perustuu mutaatioon GNPTAB-geenissä. Muita lysosomaalisia varastointitauteja tunnetaan, mutta ne perustuvat väärin syntetisoituihin hydrolaaseihin. Samankaltainen kuin I-solusairaus, myös täällä on vähentymättömien proteiinien, nukleiinihappojen ja lipidien kertymiä.
Kaikilla lysosomaalisilla varastointitauteilla on yhteistä, että johdettujen ja lysosomeista vapautuvien aineiden suhde häiriintyy purettavien aineiden kustannuksella. Lysosomeissa on todellinen ruuhka. Varastointitaudit kulkevat yleensä vakavaan suuntaan eikä niitä voida korjata syyn poistamiseksi.
Toinen riski syntyy, kun lipofiilisiä lääkkeitä otetaan heikolla pohjalla. Ne voivat kulkea lysosomien kalvojen läpi neutraalissa muodossa ulkopuolelta sisälle, mutta ei vastakkaiseen suuntaan, jos lysosomien happampi ympäristö protonoi niitä, joten lysosomotropiaa, lääkkeiden kerääntymistä lysosomeihin, voi tapahtua .Lysosomien lääkeaineiden pitoisuus voi olla 100 - 1000-kertainen veriplasman pitoisuuteen nähden.
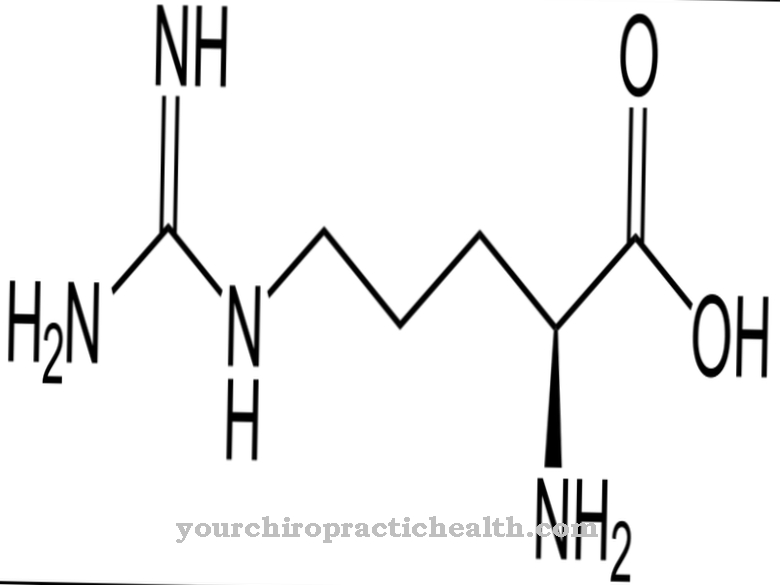

.jpg)
.jpg)







.jpg)


.jpg)