Kaikkiaan 45 erilaista lysosomaaliset varastointitaudit, jotka ovat heterogeeninen ryhmä synnynnäisiä metabolisia sairauksia. Mistä tahansa näistä sairauksista kärsivillä ihmisillä on geneettinen virhe. Kaikilla varastointitauteilla on yksi yhteinen asia: tietystä entsyymistä puuttuu tai se on vain osittain toiminnallinen.
Mikä on lysosomaalinen säilytystauti?

© designua - stock.adobe.com
Nämä synnynnäiset säilytystaudit ovat harvinaisia, koska tautia esiintyy vähemmän kuin viidellä tuhannesta ihmisestä. Eri sairauksilla on hyvin erilainen kulku, ja oireet voivat vaihdella suuresti.
Kuuluisimmat muodot lysosomaalinen varastointitauti ovat Fabryn tauti, Gaucherin tauti, Pompe-tauti ja mukopolysakkaridioosi (MPS). Niitä kutsutaan usein "lääketieteen orvoiksi", koska tie tiettyyn diagnoosiin ja sopivaan terapiaan voi olla hyvin pitkä. Joskus voi kestää vuosia, kun kärsivät saavat selville, mitä heille tapahtuu.
syyt
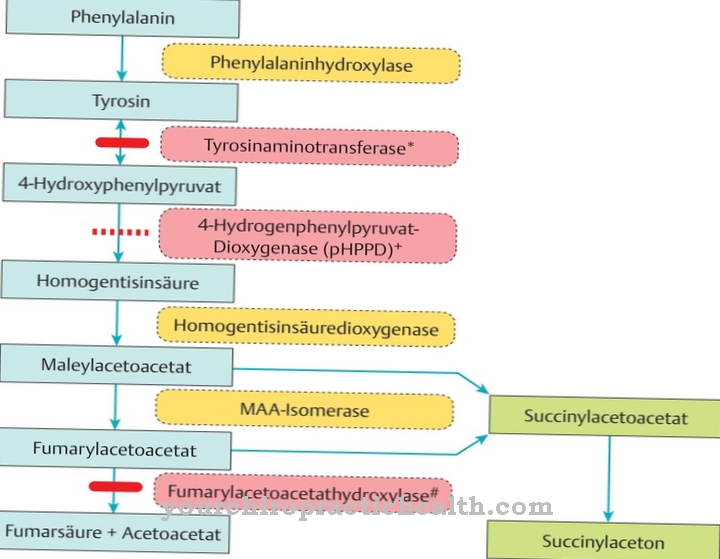
Lysosomaalisille varastointitaudeille on ominaista tietyt perinnölliset metaboliset sairaudet. Potilailta puuttuu tärkeä entsyymi, joka varmistaa metabolisen tasapainon sujuvan. Vähemmän korostuneessa muodossa tätä entsyymiä ei ainakaan ole läsnä riittävissä määrin.
Entsyymien tehtävänä on hävittää epäpuhtaudet ja jäteaineet, jotka kertyvät ihmisen organismiin aineenvaihdunnan kautta lysosomien kautta, tai prosessoida ne uudelleen siten, että oireita ei esiinny.
Entsyymipuutoksen takia tätä sujuvasti toimivaa hävittämisjaksoa ei enää taata. Haitalliset aineet asettuvat soluihin ja häiritsevät aineenvaihduntaa. Alkuvaiheessa häiriöillä ei ole huomattavaa vaikutusta, on vain muutamia rajoituksia. Kuitenkin, jos tätä aineenvaihduntahäiriötä ei hoideta entsyymipuutoksen seurauksena, oireet moninkertaistuvat, koska solut suurenevat suuresti.
Oireet, vaivat ja oireet
Pahimmassa tapauksessa nämä jäävät alle. Seurauksena on luiden, hermoston, pernan, munuaisten, lihaksen tai sydämen vaurioituminen. Fabry-tauti aiheuttaa rasvan (globotriaosyyliseramidi, Gb3) varastoinnin soluihin entsyymiaktiivisuuden vähentyneen tai poissaolon takia. Nämä ei-toivotut talletukset voivat aiheuttaa vakavaa kipua varpaissa tai sormissa, aivohalvausta ja munuaisvaurioita.
Diagnoosi ja sairauden kulku
Tämä kliininen kuva vaikuttaa samanaikaisesti erilaisiin järjestelmiin: verisuoniin, munuaisiin, sydämeen ja hermostoon. Autosomaalisesti resessiivinen Gaucherin tauti aiheuttaa "beeta-glukoserebrosidaasi" -entsyymin mutaation ja johtaa substraatin kerääntymiseen soluihin, erityisesti makrofagoihin (fagosyyteihin), jotka kuuluvat retikulo-endoteelijärjestelmään. Veren määrä muuttuu, maksa ja perna ovat laajentuneet ja luut vahingoittuvat.
Tauti on etenevä ja useimmiten etninen, koska sitä esiintyy useimmissa tapauksissa juutalaisten sukupolvien ihmisillä. Pompe-tauti tunnetaan myös nimellä "happo maltaasin puutos". Kliininen kuva kuuluu glykogeneesityypin II ryhmään. Vaikuttajilta puuttuu entsyymi "alfa-1,4-glukosidaasi" (happo maltaasi) tai sitä ei ole saatavana riittävästi. Lihasten glykogeenimäärän heikentyneen heikentymisen vuoksi potilaat kärsivät lihassolujen tuhoutumisesta sokerin varastoinnin muodossa.
Tyypin I mukopolysakkharidoosilla (MPS), joka tunnetaan myös nimellä Hunterin tauti, on useita kliinisiä syitä. Hurlerin tauti on vakavain muoto ja Scheien tauti on kliinisen patogeneesin lopussa. Näiden kahden etenemismuodon välillä on eri ominaispiirteitä. Silmiinpistävin ominaisuus on solujen lysosomeihin kertyvien hiilihydraattien heikentynyt hajoaminen.
Hunter-tautia sairastavilla potilailla voi olla lyhyt rytmi, suurennettu perna ja maksa, raskaat piirteet, paksuneutunut iho, laajentunut kieli ja hengitysvaikeudet. Lisäksi luuranko muuttuu usein lantion, selkärangan, käden luiden ja kallon alueella. Napanuorat ja [[kyynärvarvot] ovat mahdollisia.
komplikaatiot
Useimmissa tapauksissa oireet tai komplikaatiot ilmenevät hyvin myöhään tässä taudissa. Tämän vuoksi se diagnosoidaan myöhässä, jolloin varhainen hoito on useimmissa tapauksissa mahdotonta. Ilman hoitoa taudin edetessä esiintyy erilaisia valituksia ja vaurioita sisäelimiin.
Munuaiset, maksa ja perna kärsivät erityisesti. Tämä sairaus voi myös vaikuttaa sydämeen, mikä voi johtaa pahimmassa tapauksessa sydämen kuolemaan. Lisäksi munuaiset vaurioituvat ja kärsivät usein varpaiden tai sormien kipusta. Halvaus voi tapahtua myös, jos aivot ovat vaurioituneet tämän taudin vaikutuksesta. Maksa ja perna voivat suurentua ja aiheuttaa myös voimakasta kipua.
Ei ole harvinaista, että sairastuneen ihmisen luut ovat hauraita ja myös kivuliaita. Tämän taudin hoito osoittautuu vaikeaksi. Monissa tapauksissa kärsineen elinajanodote lyhenee huomattavasti. Lääkkeiden käytössä ei yleensä ole erityisiä komplikaatioita. Taudin positiivista kulkua ei kuitenkaan voida taata kaikissa tapauksissa.
Löydät lääkkeesi täältä
Pain KivulääkkeetMilloin sinun pitäisi käydä lääkärillä?
Hiusten menetys, nivelongelmat ja elinhäiriöt ovat mahdollisia merkkejä lysosomaalisesta varastointitaudista. Vierailu lääkäriin suositellaan, jos oireet toistuvat tai jos ne ilmestyvät yhtäkkiä ilman syytä. Jos oireet liittyvät aiemmin diagnosoituun entsyymivikaan tai muuhun vakavaan sairauteen, on otettava yhteys vastuulliseen lääkäriin. Hoitamaton säilytystauti voi johtaa dementiaan, hedelmättömyyteen, neuropatioihin ja muihin komplikaatioihin, joista jotkut ovat hengenvaarallisia. Siksi kaikkia mahdollisia oireita olisi tutkittava, vaikka ei ole olemassa erityistä epäilyä.
Lysosomaalisen säilytystaudin oireet voivat ilmetä vaiheittain tai kehittyä salaperäisesti, mutta vaativat aina tutkimuksen ja hoidon. Vaikuttavat ihmiset ovat parasta puhua suoraan perhelääkärilleen tai internistille. Varsinainen terapia tapahtuu yleensä sisätautien erikoistuneella klinikalla, vaikka fysioterapia tai psykoterapia voidaan yhdistää oireiden mukaan. Erityisesti terapeuttiset toimenpiteet on indikoitu sairauden usein negatiivisen kulun vuoksi.
Hoito ja hoito
Riippuen siitä, kuinka varhaisessa vaiheessa riittävä diagnoosi tehdään, näitä perinnöllisiä sairauksia voidaan hoitaa erittäin hyvin entsyymikorvaushoidolla, jotta kärsivillä ihmisillä on paljon vähemmän valituksia ja siten parempi elämänlaatu. Tätä korvaushoitoa käytetään kliinisen kuvan mukaan.
Gaucherin taudista kärsiviltä ihmisiltä puuttuu ”entsyymi ß-glukoserebrosidaasi”, jota tuotetaan bioteknologisesti ja infusoidaan potilaan kehoon. Lysosomit toimivat tehokkaasti ja kykenevät absorboimaan aineita lähiympäristöstään. Tästä syystä keinotekoisesti käytettyjä entsyymejä modifioidaan siten, että ne voidaan toimittaa lysosomeihin ihanteellisella tavalla.
Makrofaagit (fagosyytit) hajottavat soluihin kertyneet glukoserebrosidit. Tätä terapiaa voidaan verrata diabetes mellituksen insuliinihoitoon sillä erolla, että se ei ole puuttuva hormoni, vaan entsyymi, jota ei toimiteta. Keho hajottaa säännöllisesti kaikki aineet, mukaan lukien toimitettu keinotekoinen entsyymi.
Aineen tämän säännöllisen hajoamisen vuoksi potilaiden on tehtävä tämä infuusiohoito säännöllisesti elämänsä loppuun asti. Entsyymikorvaushoito ei toimi oireellisesti, vaan torjuu suoraan perinnöllisen sairauden syytä. Lääkärit kutsuvat tätä terapiaa kausaaliseksi. Hoidon periaatteita on käytettävä kaikissa neljässä edellä mainitussa yleisessä varastointitaudissa.
Pompe-potilaita hoidetaan myös infuusiohoidolla. Tässä sairaudessa toimitetaan olematonta entsyymiä "happo alfaglukosidaasi" ja se auttaa hajottamaan glykogeenia, joka on kertynyt lihaksen lysosomeihin. Lysosomaalista entsyymiä ”alfa-iduronidaasi” ei esiinny potilailla, joilla on tautityyppi ”tyypin I mukopolysakkharidoosi”, tai sitä ei ole riittävästi. Se on yksi harvimmista varastointitaudeista, joissa sokerimolekyylit kerääntyvät elimiin ja kudoksiin.
Jos prosessi on normaali, entsyymi hajottaa mukopolysakkaridit. Sokerimolekyylit ovat pitkäketjuisia ja osallistuvat tuki- ja sidekudoksen, esimerkiksi luiden, ihon, nivelnesteiden ja rustojen, kehitykseen. Jos normaali hajoamissuunta on häiritty entsyymin puuttumisen vuoksi, patologiset glykosaminoglykaanit (GAG) kertyy yksittäisiin soluihin. Tulevat terapiavaihtoehdot on suunnattu tablettien ottamiseen.
Näkymät ja ennuste
Varastointitaudin ennuste on heikko. Geneettisen sijoittelun todettiin olevan syy terveyshäiriöön. Lakivaatimukset kieltävät lääkärit ja tutkijat muuttamasta ihmisen genetiikkaa. Tästä syystä tauti pysyy elinikäisenä eikä sillä ole mahdollisuuksia toipua.
Hoitava lääkäri keskittyy esiintyvien oireiden hoitoon. Hoitamatta niitä, useat valitukset lisääntyvät ajan myötä. Luujärjestelmä on vaurioitunut ja elinten ongelmat ilmenevät. Pahimmassa tapauksessa sisäelimet toimivat väärin ja lopulta niiden toiminta epäonnistuu. Tämä uhkaa asianomaista ennenaikaisella kuolemalla.
Taudin haaste on diagnoosissa. Monilla potilailla huomionarvoisia ja voimakkaasti havaittavissa olevia valituksia esiintyy vasta myöhemmin. Seurauksena on, että geneettinen häiriö jää huomaamatta pitkään ja sairauden varhainen hoitaminen on vaikeaa. Mitä myöhemmin diagnoosi tehdään, sitä epäsuotuisampi on jatkokurssi. Taudin pitkälle edenneessä vaiheessa sisäelimet tai nivelet ovat jo vakavasti vaurioituneet. Kirurginen hoito on tarpeen, ja jos tauti etenee epäsuotuisasti, vain yksi luovuttajaelin voi pelastaa sairastuneen hengen. Varhainen hoito on siksi välttämätöntä parempaan ennusteeseen.
ennaltaehkäisy
Koska synnynnäinen geneettinen vika estää entsyymin ilmentymisen, tätä tautia ei voida hoitaa ennaltaehkäisevästi. Viimeisimmät geenitekniikan saavutukset voisivat kuitenkin tarjota lähestymistavan tällä alalla.
Jälkihoito
Tämän taudin takia ihmiset kärsivät monista erilaisista komplikaatioista ja vaivoista. Näillä kaikilla on pääsääntöisesti erittäin kielteinen vaikutus kärsineiden elämänlaatuun, joten diagnoosi olisi tehtävä hyvin varhaisessa vaiheessa.Mitä aikaisemmin lääkäriin neuvotellaan, sitä parempi tämän taudin eteneminen yleensä on.
Tämän taudin vakavuus voi olla hyvin erilainen, joten yleinen ennustaminen ei usein ole mahdollista. Vaikuttavat kärsivät vakavista vaurioista sisäelimissä. Munuaiset ja sydän kärsivät ensisijaisesti, joten lapsi voi kuolla muutaman ensimmäisen päivän aikana, jos oireita ei korjata ajoissa. Rasvaa on myös kehon eri osissa.
Erityisesti vahingoitetaan sormia ja varpaita, mikä voi vähentää merkittävästi estetiikkaa kyseessä olevalle henkilölle. Yleensä munuaiset ja aivot vaurioituvat jatkovaiheessa, joten kyseinen henkilö kuolee tämän vaurion seurauksena. Vanhemmat ja sukulaiset kärsivät myös usein masennuksesta tai muista mielenterveyden häiriöistä sairauden vuoksi.
Voit tehdä sen itse
Lysosomaaliset varastointitaudit vaativat usein intensiivistä lääketieteellistä hoitoa. Usein ei ole tarpeeksi mahdollisuuksia itseapuun. Vaikutuksen alaisten lasten vanhemmat kokevat usein vakavaa stressiä kotiympäristössään, koska heidän lapsensa tarvitsevat jatkuvaa hoitoa ja huomiota.
Kliiniset kuvat yksittäisistä varastointitaudeista ovat erilaisia. On sekä helppoja että erittäin vaikeita muotoja. Yksi esimerkki on Gaucherin tauti. Vanhempien apu rajoittuu usein vakavasti vammaisen lapsen ruokintaan. Lievemmissä tapauksissa elinajanodote voi olla melkein normaali. Tästä huolimatta jatkuva lääketieteellinen valvonta on välttämätöntä mahdollisten komplikaatioiden välttämiseksi. Säännöllinen fyysinen aktiivisuus on yksi oheishoidoista, jotka voidaan suorittaa myös kotona. Lisäksi on järjestettävä huolellinen syöpäseulontakoe. Se vaatii jatkuvia käyntejä lääkäriltä lapsensa kanssa vanhemmilta. Sama pätee muihin lysosomaalisiin varastointitauteihin.
Joissakin sairauksissa fyysisten vammojen lisäksi voi ilmetä myös psyykkisiä vammoja, jotka vaativat edelleen erityistä tukea. Tiettyjen sairauksien, kuten Hunterin taudin, lievemmissä muodoissa esiintyy aluksi vain luuston muutoksia ja kasvojen dysmorfismeja. Täällä kuitenkin kärsivä potilas pystyy usein elämään itsenäisen elämän. Tässä yhteydessä tarvitaan kuitenkin myös jatkuvia lääketieteellisiä tutkimuksia mahdollisten komplikaatioiden, kuten sydämen vajaatoiminnan tai hengityselinsairauksien, sulkemiseksi pois. Potilas pystyy käsittelemään fyysisten muodonmuutosten aiheuttamaa psykologista stressiä psykologisen ohjauksen avulla.
.jpg)
.jpg)

.jpg)






.jpg)